Discovery of new molecules and materials with desired properties is a practical goal of chemical research. A promising way to significantly accelerate the latter process is to incorporate all available knowledge and data to plan the synthesis of the next materials. Our group focuses on developing computational methods and algorithms to discover new materials for a wide spectrum of applications 10x faster using the state-of-the-art machine learning algorithms and artificial intelligence to go beyond the human intuitions. We predict the properties of chemicals and materials, explore the chemical space for inverse design, and predict the synthesis pathways ahead of experiments based on data. These efforts are complemented by the quantum mechanical and molecular mechanical simulations that provide atomistic snapshots of what is happening at the microscopic scale in the materials. Our current research interests can be categorized into four areas broadly defined as 1) developing AI methods for novel chemical discovery, 2) developing AI methods for automated chemical synthesis, 3) atomic-scale understanding and designing of functional molecules and materials, and 4) developing fast and accurate electronic structure methods that can treat large molecules and materials more reliably.
Discovery of new molecules and materials with desired properties is a practical goal of chemical research. A promising way to significantly accelerate the latter process is to incorporate all available knowledge and data to plan the synthesis of the next materials. Our group focuses on developing computational methods and algorithms to discover new materials for a wide spectrum of applications 10x faster using the state-of-the-art machine learning algorithms and artificial intelligence to go beyond the human intuitions. We predict the properties of chemicals and materials, explore the chemical space for inverse design, and predict the synthesis pathways ahead of experiments based on data. These efforts are complemented by the quantum mechanical and molecular mechanical simulations that provide atomistic snapshots of what is happening at the microscopic scale in the materials. Our current research interests can be categorized into four areas broadly defined as 1) developing AI methods for novel chemical discovery, 2) developing AI methods for automated chemical synthesis, 3) atomic-scale understanding and designing of functional molecules and materials, and 4) developing fast and accurate electronic structure methods that can treat large molecules and materials more reliably.
AI for Chemical Discovery
AI for Chemical Discovery
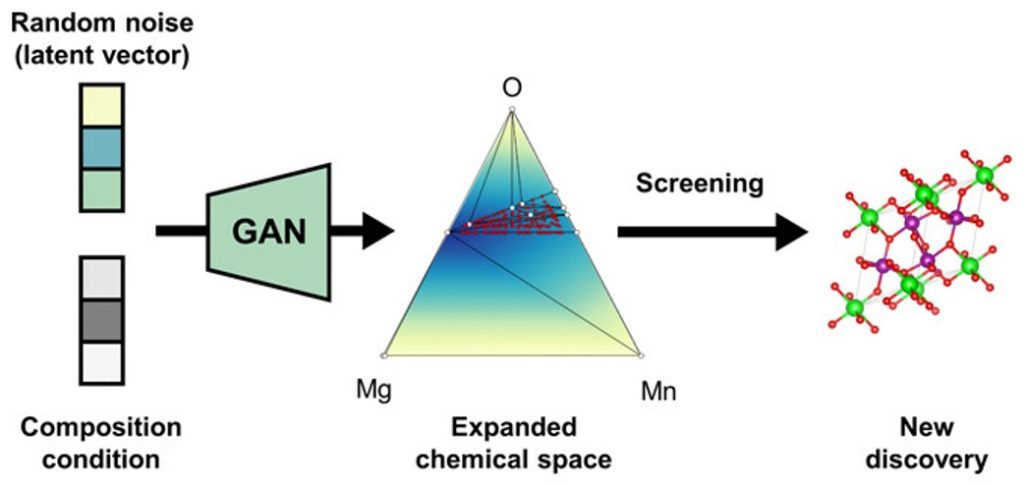
Developing high-performance advanced materials requires a deeper insight and search into the chemical space. Until recently, exploration of materials space using chemical intuitions built upon existing materials has been the general strategy, but this direct design approach is often time and resource consuming and poses a significant bottleneck to solve the materials challenges of future sustainability in a timely manner. To accelerate this conventional design process, inverse design, which outputs materials with pre-defined target properties, has emerged as a significant materials informatics platform in recent years by leveraging hidden knowledge obtained from materials data. These include AI-based property predictions that can facilitate fast screening of the database for different applications, or inverse design based on generative models.
Developing high-performance advanced materials requires a deeper insight and search into the chemical space. Until recently, exploration of materials space using chemical intuitions built upon existing materials has been the general strategy, but this direct design approach is often time and resource consuming and poses a significant bottleneck to solve the materials challenges of future sustainability in a timely manner. To accelerate this conventional design process, inverse design, which outputs materials with pre-defined target properties, has emerged as a significant materials informatics platform in recent years by leveraging hidden knowledge obtained from materials data. These include AI-based property predictions that can facilitate fast screening of the database for different applications, or inverse design based on generative models.
AI for Synthesis
AI for Synthesis
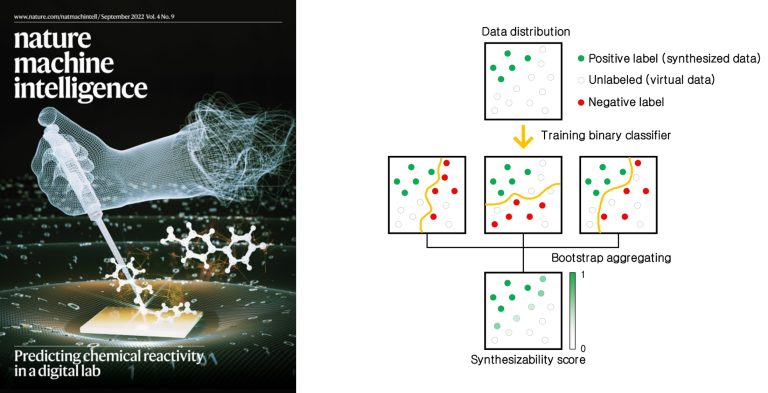
One great challenge in digital discovery is that many of the molecules and materials that are computationally designed are often discarded in the laboratories since they are not synthesizable. There are some widely employed approximate approaches to estimate synthesizability, such as the thermodynamic stability, but they could produce too many potentially synthesizable candidates or miss important metastable materials. The synthesis being a complex phenomenon, simple thermodynamic stability alone or other heuristic rules may not be able to yield reliable prediction capability. Our work in this area is related to predict the materials synthesizability, molecular and inorganic retrosynthesis, organic reactivity prediction, and synthesis-aware molecule generation. Automating the process of synthesis by artificial intelligence would accelerate synthesis design in future digital laboratories.
One great challenge in digital discovery is that many of the molecules and materials that are computationally designed are often discarded in the laboratories since they are not synthesizable. There are some widely employed approximate approaches to estimate synthesizability, such as the thermodynamic stability, but they could produce too many potentially synthesizable candidates or miss important metastable materials. The synthesis being a complex phenomenon, simple thermodynamic stability alone or other heuristic rules may not be able to yield reliable prediction capability. Our work in this area is related to predict the materials synthesizability, molecular and inorganic retrosynthesis, organic reactivity prediction, and synthesis-aware molecule generation. Automating the process of synthesis by artificial intelligence would accelerate synthesis design in future digital laboratories.
Materials Understanding
Materials Understanding
Despite exciting experimental progresses in synthesizing, characterizing, and visualizing the materials systems in a microscopic scale, obtaining full atomistic details and underlying mechanisms of many intriguing materials phenomena are still very challenging, making the directed materials design difficult. Computational modeling based on quantum mechanics and molecular mechanics offer significant opportunities to address these challenges by providing atomistic level behaviors of molecules and materials. Muliscale and statistical techniques can further bridge the gap between microscopic understanding and macroscopic experimental measurements. We aim to reveal materials design guidelines by the latter understanding and perform computational high-throughput virtual screening to rapidly explore the large chemical space in a systematic fashion, identifying promising materials candidates that can be tested experimentally
Despite exciting experimental progresses in synthesizing, characterizing, and visualizing the materials systems in a microscopic scale, obtaining full atomistic details and underlying mechanisms of many intriguing materials phenomena are still very challenging, making the directed materials design difficult. Computational modeling based on quantum mechanics and molecular mechanics offer significant opportunities to address these challenges by providing atomistic level behaviors of molecules and materials. Muliscale and statistical techniques can further bridge the gap between microscopic understanding and macroscopic experimental measurements. We aim to reveal materials design guidelines by the latter understanding and perform computational high-throughput virtual screening to rapidly explore the large chemical space in a systematic fashion, identifying promising materials candidates that can be tested experimentally
Electronic structure methods
Electronic structure methods
Accuracy and feasibility of modern computational modelling techniques based on quantum mechanics has progressed recently to a level that allows one to make reliable predictions ahead of experiments. However, these methods, such as those based on density functional theory, still scale unfavorably to treat large systems, or their accuracy is well-known to be insufficient for certain molecular or materials properties compared to correlated wavefunction-based methods. Our group develops new density functional methods that are fast and accurate to accelerate the computational material discovery even further.
Accuracy and feasibility of modern computational modelling techniques based on quantum mechanics has progressed recently to a level that allows one to make reliable predictions ahead of experiments. However, these methods, such as those based on density functional theory, still scale unfavorably to treat large systems, or their accuracy is well-known to be insufficient for certain molecular or materials properties compared to correlated wavefunction-based methods. Our group develops new density functional methods that are fast and accurate to accelerate the computational material discovery even further.